Recombinant Human Iduronate 2-sulfatase (IDS)
-
中文名稱(chēng):人IDS重組蛋白
-
貨號(hào):CSB-YP010998HU
-
規(guī)格:
-
來(lái)源:Yeast
-
其他:
-
中文名稱(chēng):人IDS重組蛋白
-
貨號(hào):CSB-EP010998HU
-
規(guī)格:
-
來(lái)源:E.coli
-
其他:
-
中文名稱(chēng):人IDS重組蛋白
-
貨號(hào):CSB-EP010998HU-B
-
規(guī)格:
-
來(lái)源:E.coli
-
共軛:Avi-tag Biotinylated
E. coli biotin ligase (BirA) is highly specific in covalently attaching biotin to the 15 amino acid AviTag peptide. This recombinant protein was biotinylated in vivo by AviTag-BirA technology, which method is BriA catalyzes amide linkage between the biotin and the specific lysine of the AviTag.
-
其他:
-
中文名稱(chēng):人IDS重組蛋白
-
貨號(hào):CSB-BP010998HU
-
規(guī)格:
-
來(lái)源:Baculovirus
-
其他:
-
中文名稱(chēng):人IDS重組蛋白
-
貨號(hào):CSB-MP010998HU
-
規(guī)格:
-
來(lái)源:Mammalian cell
-
其他:
產(chǎn)品詳情
-
純度:>85% (SDS-PAGE)
-
基因名:
-
Uniprot No.:
-
別名:Alpha L iduronate sulfate sulfatase; Alpha-L-iduronate sulfate sulfatase; AW214631; Ids; IDS_HUMAN; Iduronate 2 sulfatase 14 kDa chain; Iduronate 2 sulfatase 42 kDa chain; Iduronate 2 sulfatase; Iduronate 2-sulfatase 14 kDa chain; Iduronate sulfatase; Idursulfase; MPS2; RP23-29M4.1; SIDS
-
種屬:Homo sapiens (Human)
-
蛋白長(zhǎng)度:Full Length of Mature Protein
-
表達(dá)區(qū)域:34-455
-
氨基酸序列TDALNVL LIIVDDLRPS LGCYGDKLVR SPNIDQLASH SLLFQNAFAQ QAVCAPSRVS FLTGRRPDTT RLYDFNSYWR VHAGNFSTIP QYFKENGYVT MSVGKVFHPG ISSNHTDDSP YSWSFPPYHP SSEKYENTKT CRGPDGELHA NLLCPVDVLD VPEGTLPDKQ STEQAIQLLE KMKTSASPFF LAVGYHKPHI PFRYPKEFQK LYPLENITLA PDPEVPDGLP PVAYNPWMDI RQREDVQALN ISVPYGPIPV DFQRKIRQSY FASVSYLDTQ VGRLLSALDD LQLANSTIIA FTSDHGWALG EHGEWAKYSN FDVATHVPLI FYVPGRTASL PEAGEKLFPY LDPFDSASQL MEPGRQSMDL VELVSLFPTL AGLAGLQVPP RCPVPSFHVE LCREGKNLLK HFRFRDLEED PYLPG
-
蛋白標(biāo)簽:Tag?type?will?be?determined?during?the?manufacturing?process.
The tag type will be determined during production process. If you have specified tag type, please tell us and we will develop the specified tag preferentially. -
產(chǎn)品提供形式:Lyophilized powder
Note: We will preferentially ship the format that we have in stock, however, if you have any special requirement for the format, please remark your requirement when placing the order, we will prepare according to your demand. -
復(fù)溶:We recommend that this vial be briefly centrifuged prior to opening to bring the contents to the bottom. Please reconstitute protein in deionized sterile water to a concentration of 0.1-1.0 mg/mL.We recommend to add 5-50% of glycerol (final concentration) and aliquot for long-term storage at -20℃/-80℃. Our default final concentration of glycerol is 50%. Customers could use it as reference.
-
儲(chǔ)存條件:Store at -20°C/-80°C upon receipt, aliquoting is necessary for mutiple use. Avoid repeated freeze-thaw cycles.
-
保質(zhì)期:The shelf life is related to many factors, storage state, buffer ingredients, storage temperature and the stability of the protein itself.
Generally, the shelf life of liquid form is 6 months at -20°C/-80°C. The shelf life of lyophilized form is 12 months at -20°C/-80°C. -
貨期:Delivery time may differ from different purchasing way or location, please kindly consult your local distributors for specific delivery time.Note: All of our proteins are default shipped with normal blue ice packs, if you request to ship with dry ice, please communicate with us in advance and extra fees will be charged.
-
注意事項(xiàng):Repeated freezing and thawing is not recommended. Store working aliquots at 4°C for up to one week.
-
Datasheet :Please contact us to get it.
產(chǎn)品評(píng)價(jià)

相關(guān)產(chǎn)品
靶點(diǎn)詳情
-
功能:Lysosomal enzyme involved in the degradation pathway of dermatan sulfate and heparan sulfate.
-
基因功能參考文獻(xiàn):
- IDS structure revealed by X-ray crystallography provides essential insight into multiple mechanisms by which pathogenic mutations interfere with enzyme function, and a compelling explanation for severe Hunter syndrome phenotypes. PMID: 28593992
- Study analyzed the genotype-phenotype relationship for 17 patients with mucopolysaccharidosis II and performed expression studies for 12 variants, nine of which have not been reported previously; speculated that very low or cell-type-specific IDS residual activity is sufficient to prevent the neuronal phenotype. PMID: 28543354
- Study identified 16 novel mutations in the IDS gene and revealed that the severe type of mucopolysaccharidosis type II is strongly associated with large structural alteration of the gene. PMID: 27246110
- Functional characterization of all the novel sequence variants identified in the study would be helpful to confirm the clinical significance and to determine the effect of these variations on the function of respective proteins (IDUA and IDS) PMID: 27146977
- A splicing mutation, c.709-1G>A, was detected in the proband, for which his mother was heterozygous. PMID: 28186595
- Extensive iduronate 2-sulfatase (Hunter syndrome) (IDS) gene deletions were identified in four mucopolysaccharidosis type II (MPSII) patients. PMID: 26762690
- Two new mutations were discovered: p.K236N (c.708G>C) and p.Q80K (c.238C>A) which resulted in a severe phenotype and early death of Muccopolysaccharridosis Type II patients from Bulgaria and Macedonia. PMID: 22286622
- p.Ser142Phe and p.Ile360Tyrfs*31 mutations caused the severe disease manifestation PMID: 24780617
- This study evaluated a novel mutation in the IDS gene among 8 male Hunter syndrome patients; there was a quantitative deficiency of NK and B cell with normal responses in other immune parameters. PMID: 25038527
- 30 novel iduronate sulfatase mutations have been identified in mucopolysaccharidosis type II Latin American patients. PMID: 24125893
- Identification of a splice site mutation in the IDS gene associated with mucopolysaccharidosis type II. PMID: 23867855
- a novel (p.R468P) and five known (p.R88C, p.D148V, p.G224A, p.Y348X, and p.R468Q) IDS mutations were shown to result in proteins with little or no IDS activity and altered protein processing, when expressed in COS7 cells PMID: 22990955
- A report of a novel IDS nonsense mutation resulting in mucopolysaccharidosis type II in several patients from a Chinese family. PMID: 22622771
- genetically analyze patients with severe Hunter syndrome that showed a total deletion of the iduronate-2-sulphatase (IDS) gene PMID: 22492741
- Family members with 3 generations of X-inactivation with Hunter syndrome have 1568A>G missence mutation in the IDS gene PMID: 21062272
- LCR-initiated rearrangements at the IDS locus, completed with Alu-mediated recombination or non-homologous end joining PMID: 21593745
- Hunter syndrome in Thailand is caused by a diverse set of defects affecting both IDS protein production and activity. PMID: 18500569
- study describes a woman with mild manifestations of Hunter syndrome who gave birth to a daughter; both the mother and daughter carried the p.R443X mutation in exon 9 of the ID2S gene PMID: 21108396
- The in vivo correction of heritable gene lesions at the RNA level operating via a correction mechanism akin to RNA-editing, was observed for IDS mutant transcript. PMID: 20104590
- The results illustrated that the deletion and frame-shift mutation of c.876-877 del TC detected in IDS gene was a novel pathologic mutation,, which was the underlying cause of MPS II of this patient. PMID: 19933090
- patterns of cytosine methylation in the entire IDS gene PMID: 15146464
- Mucopolysaccharidosis type II patients with sever CNS involvement and age of onset by 3 years of age had four IDS amino acid substitutions S333L,C53X,E341K, and P480R. PMID: 15500445
- a total of 17 identified missense, small deletion, and nonsense mutations were further characterized by transient expression studies. PMID: 15614569
- large deletion correlated with the severe phenotype of this Hunter syndrome patient. PMID: 15909065
- The IDS gene was analyzed in Japanese patients with mucopolysaccharidosis II. PMID: 16133661
- IDS activity in female carriers was less than a half of the normal level PMID: 16480701
- These findings suggest methylation patterns in the beginning of IDS genomic region are polymorphic in humans and that hypermethylation in this region in some individuals predisposes them to CpG mutations resulting in Hunter syndrome. PMID: 16617305
- The balance between constitutive and cryptic splice sites in the IDS gene is very sensitive. PMID: 16699754
- A new point mutation (T1140C) in exon 8 of the IDS gene was found in Hunter syndrome. PMID: 16735228
- the IDS gene is prone to splicing mutations in Portuguese patients with mucopolysaccharidosis type II PMID: 17063374
- analysis of iduronate-2-sulfatase enzymatic activity, protein processing and structure PMID: 17091340
- Two novel mutations were identified in the human iduronate-2-sulfatase (IDS) gene in two patients from unrelated families with mucopolysaccharidosis type II(MPS II). PMID: 17284421
- Identification of a novel nonsense mutation (p.Y54X) in the IDS gene of severely affected MPS II patients of African origin. PMID: 17616540
- The molecular characterization of one novel missense mutation (p.S305P) and 1 splice site mutation (c.1006 +5G > C) associated with mucopolysaccharidosis type II was presented. PMID: 17655837
- frame-shift deletion mutation (1062 del 16) was identified in exon 7 of the patient's IDS gene PMID: 17657858
- A new mutation, an A>T change at nucleotide 595, substitutes a premature stop codon for a lysine at amino acid 199 of the IDS enzyme. PMID: 18546295
- IDS has a role in glucose-stimulated insulin secretion via a mechanism that involves the activation of exocytosis through phosphorylation of PKCalpha and MARCKS. PMID: 19602578
- novel mutations in Italian patients with mucopolysaccharidosis type II PMID: 11462244
顯示更多
收起更多
-
相關(guān)疾病:Mucopolysaccharidosis 2 (MPS2)
-
亞細(xì)胞定位:Lysosome.
-
蛋白家族:Sulfatase family
-
組織特異性:Liver, kidney, lung, and placenta.
-
數(shù)據(jù)庫(kù)鏈接:
Most popular with customers
-
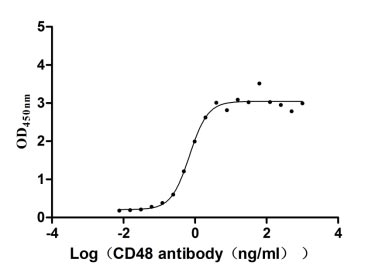
Recombinant Human CD48 antigen (CD48) (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
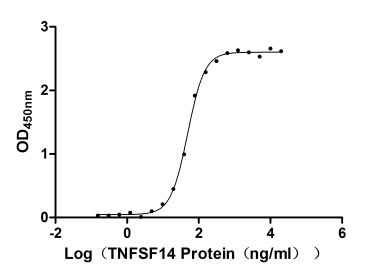
Recombinant Human Tumor necrosis factor ligand superfamily member 14 (TNFSF14), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
-AC1.jpg)
Recombinant Human Pro-neuregulin-1, membrane-bound isoform (NRG1), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
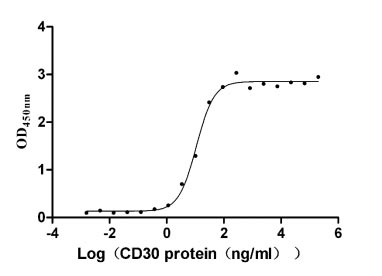
Recombinant Human Tumor necrosis factor ligand superfamily member 8 (TNFSF8), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
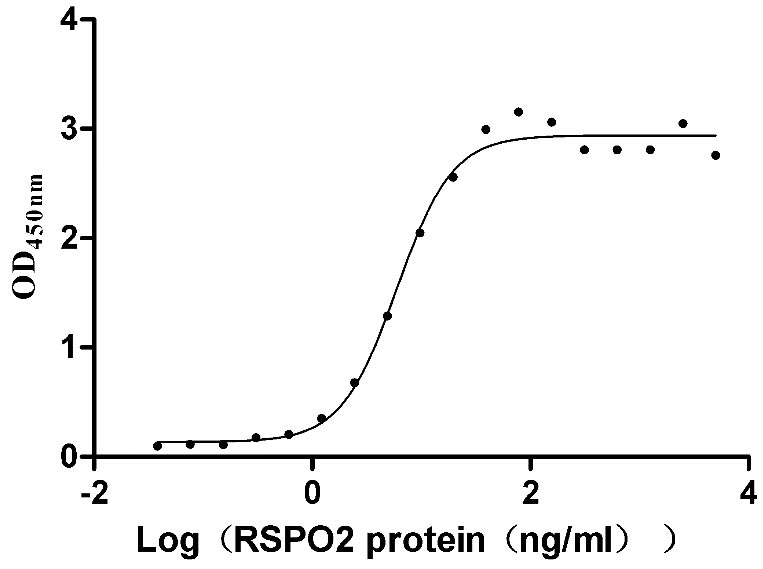
Recombinant Human E3 ubiquitin-protein ligase ZNRF3 (ZNRF3), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
Recombinant Human Claudin-4 (CLDN4)-VLPs (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
-AC1.jpg)
Recombinant Human Dickkopf-related protein 1 (DKK1) (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
Recombinant Human Interleukin-2 receptor subunit alpha (IL2RA), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)





")
")






